早期発症型
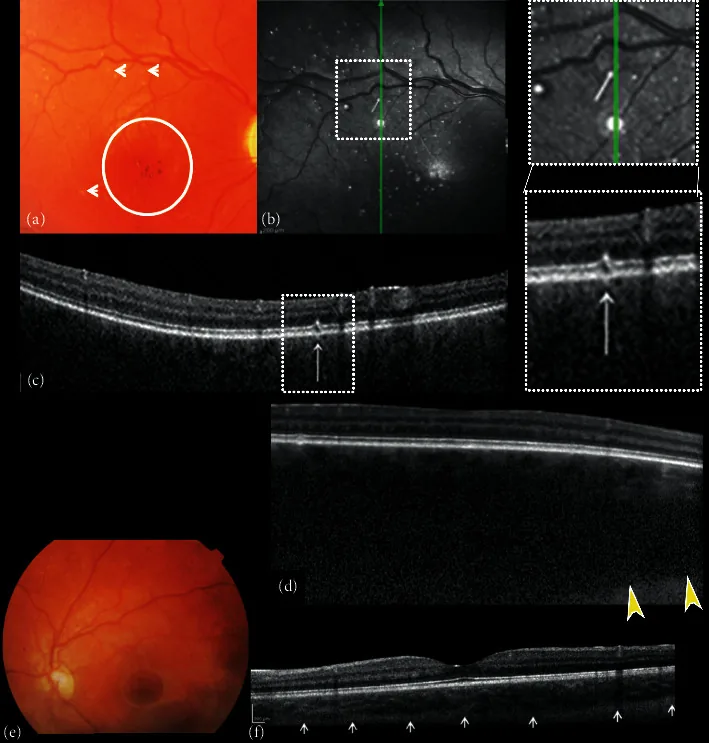
黄斑症:最も特徴的な所見。生後35日から出現しうる。
牛眼様黄斑症:中心部脈絡網膜萎縮と色素環を呈する特徴的パターン。
網膜色素変性様変化:広範な網膜変性が生じる。
視神経萎縮:進行例で認められる。
眼底蛍光造影(FAF):中心窩低自発蛍光・境界部過自発蛍光パターン。

コバラミンC欠乏症(cblC型)は、細胞内ビタミンB12(コバラミン)代謝の先天代謝異常症である。正式名称はメチルマロン酸尿症およびホモシスチン尿症cblC型(OMIM #277400)。cbl代謝異常症全体の約80%を占める最多型である。
遺伝形式は常染色体劣性。原因はMMACHC遺伝子(1p34.2)の変異である。MMACHCタンパクは細胞内コバラミン代謝の初期段階を担い、その機能喪失によりアデノシルコバラミン(AdoCbl)およびメチルコバラミン(MeCbl)の両方の合成が障害される。
推定発生率は1:100,000〜1:200,000とされるが3)4)、中国山東省の調査では1:3,920と高い頻度が報告されている。米国では2000年代初頭より新生児スクリーニング(NBS)の対象となっている4)。
発症年齢によって早期発症型(生後1年以内、全体の86〜88%)と遅発型(1歳以降)に大別される。遅発型の報告は160例以上、成人発症(18歳以上)は30例以上が報告されている3)。
血中C3プロピオニルカルニチン高値を指標としたNBSにより早期発見が可能である。ただし、遅発型や軽症型ではNBSをすり抜ける例があり4)、スクリーニング正常でも臨床的に疑わしい場合は血漿Hcy・尿中MMAの追加検査が必要である。
発症年齢により臨床像が大きく異なる。
早期発症型の眼症状:
遅発型の主症状(眼外):
早期発症型と遅発型では眼底所見が大きく異なる。
早期発症型
黄斑症:最も特徴的な所見。生後35日から出現しうる。
牛眼様黄斑症:中心部脈絡網膜萎縮と色素環を呈する特徴的パターン。
網膜色素変性様変化:広範な網膜変性が生じる。
視神経萎縮:進行例で認められる。
眼底蛍光造影(FAF):中心窩低自発蛍光・境界部過自発蛍光パターン。
遅発型
眼底正常例が多い:重篤な眼所見を呈しないことが多い3)4)。
脊髄MRI所見:後索の高信号病変が特徴的4)。
脳MRI白質病変:一部の遅発型で認められる3)。
高血圧性網膜症:腎合併症(血栓性微小血管障害)を伴う重症例で生じうる2)。
全身合併症として代謝性アシドーシス・筋緊張低下・小頭症・心筋症(50%)1)・腎血栓性微小血管障害(TMA)2)が知られる。
遅発型では早期発症型のような重篤な黄斑症・網膜変性は稀で、眼底正常例が多い3)4)。ただし腎合併症を伴う重症例では高血圧性網膜症を呈することがある2)。神経症状の改善は治療反応良好であることが多いが、視機能障害が残存した例も報告されている3)。
コバラミンC欠乏症はMMACHC遺伝子の変異による常染色体劣性遺伝疾患である。80以上の変異が報告されており、変異の種類により表現型が大きく異なる。
主要な変異の頻度と対応する表現型を以下に示す。
| 変異 | 頻度・系統 | 主な表現型 |
|---|---|---|
| c.271dupA | 40〜61%・欧州系 | 早期発症・最重症 |
| c.394C>T | 約20% | 遅発型 |
| c.609G>A | 48〜55%・東アジア系 | 乳児〜小児 |
その他の主要変異として:
なお、PRDX1遺伝子変異によるMMACHCのエピジェネティックサイレンシングが原因となる「epi-cblC」も存在し、二次性のcblC様病態を引き起こす。
代謝異常を示す検査所見が診断の中心となる。
| 検査項目 | 典型的所見 | 意義 |
|---|---|---|
| 血漿Hcy | 著明高値 | スクリーニング |
| 尿中MMA | 高値 | 代謝ブロック確認 |
| 遺伝子検査 | MMACHC変異 | 確定診断 |
報告された具体的数値として:血漿Hcy 101.5〜1700.5 µmol/L2)3)、尿中MMA 85〜802 mmol/mol3)4)。血清ビタミンB12は遅発型では正常範囲のことが多い(448〜625 pg/mL)3)4)。
食事性B12欠乏・悪性貧血・他のcbl代謝異常(cblD・cblF等)・MTHFR欠乏症との鑑別が重要3)4)。Hcy高値とMMA高値の組み合わせはcblCに特徴的であり、Hcy高値のみ(MMA正常)はMTHFR欠乏等を示唆する。
ある。遅発型cblCでは血清B12が正常範囲にあることが多く3)4)、B12正常のみで否定してはならない。神経症状・精神症状がある若年患者では血漿HcyとMMAを必ず測定し、高値であれば遺伝子検査でMMACHCを解析する必要がある。
治療の主軸はヒドロキシコバラミン(OH-Cbl)の非経口投与であり、ベタイン・フォリン酸・L-カルニチンを組み合わせる多剤併用療法が行われる。
非経口投与が基本。投与量の目安:1 mg/日または0.3 mg/kg/日の筋肉内注射(IM)4)。報告された投与量:
Aillietら(2022)の遅発型成人例では25 mg/日の皮下注射(SC)を施行し、神経症状の改善を得た3)。Goyneら(2023)の症例では5 mg/日IMを開始した4)。Hjalmarssонら(2024)の重症心不全合併例では30 mg/日の静脈内投与(IV)が行われた1)。
ホモシステインのメチオニンへの再メチル化を促進する。投与量の目安:250 mg/kg/日を3分割4)。報告された投与量:100 mg/kg/日2)、1 g×3回4)、2〜3 g×3回1)。
治療目標はMMAおよびHcyの低下と代謝指標の正常化である。適切な治療によりHcyの著明な低下が得られる:
Akarら(2024)の腎TMA合併重症例では、治療開始後にHcyが1,700.5から4.6 µmol/Lへと正常化した2)。Hjalmarssонら(2024)の心筋症合併例では、MMAが97から8.1 mmol/mol・Hcyが91から31.8 µmol/Lへ改善した1)。
遅発型では治療反応が比較的良好であるが4)、視機能回復は限定的な場合がある3)。早期発症型では適切な治療を行っても10代で法的盲に至る例が多い。
早期発症型では、代謝指標が改善しても黄斑症・網膜変性の視機能予後は不良であることが多く、10代で法的盲に至る例が報告されている。遅発型では神経症状への治療反応は比較的良好だが、残存する視機能障害は限定的にしか回復しない3)。早期診断・早期治療開始が最善の予後をもたらす可能性がある。
MMACHCタンパクはリソソームにおいてコバラミン-ハプトコリン(R)複合体と結合し、コバラミンからR基を切り離して細胞内で利用可能な共通コバラミン中間体を生成する。この中間体はその後2つの経路に分岐する。
MMACHC機能喪失により両経路が障害されると:
眼組織への影響としては、メチオニン低下によるグルタチオン(GSH)合成障害が網膜色素上皮(RPE)の酸化ストレス防御を低下させ、黄斑変性の一因となると考えられている。高Hcyによる血管内皮障害が網膜・脈絡膜循環を障害する機序も推察されている。
Hjalmarssонら(2024)は、心筋症を合併したcblC患者においてLVAD(左室補助装置)植え込みと続く心臓移植を施行した世界初の症例を報告した1)。高用量OH-Cbl(30 mg/日IV)・ベタイン・フォリン酸・L-カルニチンの代謝治療と組み合わせ、代謝指標の改善(MMA 97→8.1、Hcy 91→31.8)とともに良好な転帰が得られた。
心臓移植後もcblCに対する代謝治療の継続が不可欠であることが示された1)。
遅発型・軽症型ではNBSで見逃される例があることが示されており4)、神経症状・精神症状を呈する若年成人においてB12正常でもHcyとMMAを測定することの重要性が強調されている3)4)。
PRDX1遺伝子変異によるMMACHCのエピジェネティックサイレンシングで生じる「epi-cblC」が報告されている。遺伝子変異がない場合でもcblC様病態を呈しうることから、診断アルゴリズムの再考が求められている。
Aillietら(2022)は遅発型cblCの家族スクリーニングにより無症候性ホモ接合体を早期発見した経験を報告し、プロバンドの診断を契機とした積極的な家族スクリーニングの意義を強調した3)。