眼皮膚白皮症(OCA)
常染色体劣性遺伝:最も多い群。皮膚・毛髪・眼すべてに色素欠乏を生じる。
8遺伝子・8サブタイプ:OCA1(TYR)・OCA2(OCA2/P遺伝子)・OCA3(TYRP1)・OCA4(SLC45A2)・OCA8(DCT)など。
有病率:世界で約1/17,000。白人ではOCA1が約50%を占め、アフリカ系ではOCA2が最多(1/10,000)1)。中国漢民族ではOCA1が70.1%、OCA2が10.2%1)。

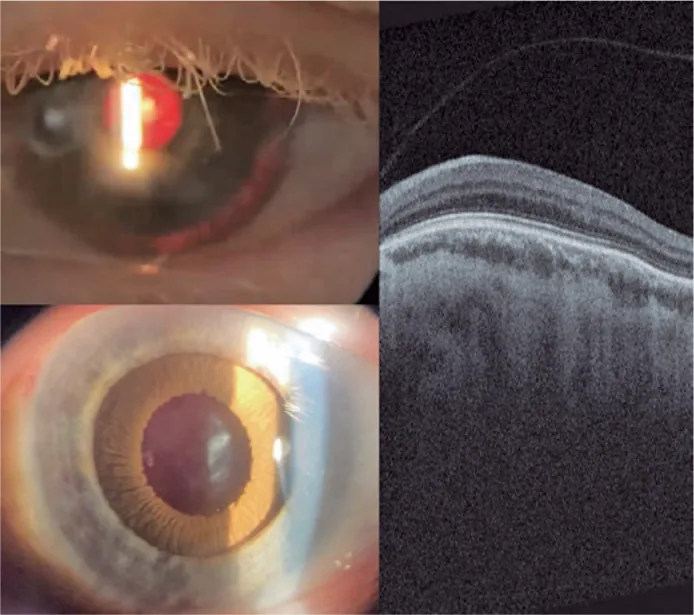
白皮症(Albinism)は、メラニン色素の生合成または輸送に関わる遺伝子変異により、皮膚・毛髪・眼の色素が欠如または減少する遺伝性疾患群である。色素欠乏に加え、中心窩形成不全・眼振・視路異常交叉などの特有の眼・視路異常を伴う点が本疾患群の特徴である。
根治的治療はなく、視機能障害は非進行性(静止性)である。日本では指定難病(難病法)に指定されている。
白皮症は大きく3群に分類される。
眼皮膚白皮症(OCA)
常染色体劣性遺伝:最も多い群。皮膚・毛髪・眼すべてに色素欠乏を生じる。
8遺伝子・8サブタイプ:OCA1(TYR)・OCA2(OCA2/P遺伝子)・OCA3(TYRP1)・OCA4(SLC45A2)・OCA8(DCT)など。
有病率:世界で約1/17,000。白人ではOCA1が約50%を占め、アフリカ系ではOCA2が最多(1/10,000)1)。中国漢民族ではOCA1が70.1%、OCA2が10.2%1)。
眼白皮症(OA)
X連鎖劣性遺伝:男性に発症。皮膚・毛髪の色素は正常または軽度低下にとどまり、眼所見が主体。
GPR143遺伝子変異:メラノソーム生合成シグナル伝達に関与。192変異が報告されている3)。
保因者女性:眼底に泥はね様モザイク状低色素パターンを示す。約80%に網膜色素異常が認められる3)。
症候群性白皮症
Hermansky-Pudlak症候群(HPS):OCA+血小板機能異常(出血傾向)+LRO生合成障害。11サブタイプが存在する4)5)。肺線維症・炎症性腸疾患を合併しうる。
Chediak-Higashi症候群(CHS):OCA+免疫不全+神経障害。LYST遺伝子変異。
12遺伝子:症候群性白皮症は12遺伝子が同定されている2)。
世界有病率は約1/17,000であり1)、欧州では約1/12,000と報告されている2)。日本人ではOCA1が34%・OCA4が27%・HPS1が10%の分布が知られている。
OCAは常染色体劣性遺伝であり、両親がともに変異アレルを1つずつ保有する保因者の場合、25%の確率で子に発症する。両親が表現型上正常でも発症しうるため、家族歴がなくても珍しくない。OAはX連鎖劣性遺伝で主に男性が発症し、母親が保因者となる。遺伝子検査と遺伝カウンセリングが重要である。
主なサブタイプ別の視力目安を以下に示す。
| サブタイプ | 視力範囲の目安 |
|---|---|
| OCA1 | 0.1以下が多い |
| OCA2/OCA4 | 0.1〜0.3 |
| OCA8 | 0.1〜0.4(LogMAR)2) |
| OA1 | 0.1〜0.4 |
| HPS-11 | 20/200程度5) |
白皮症による視機能障害は静止性(非進行性)であり、加齢とともに悪化することはない。ただし、適切な屈折矯正や弱視治療が行われなかった場合には視力が改善しないまま固定されることがある。屈折矯正・ロービジョンケアの早期介入が重要である。
各サブタイプの主な原因遺伝子と特徴を以下に示す。
| サブタイプ | 原因遺伝子 | 主な特徴 |
|---|---|---|
| OCA1 | TYR | チロシナーゼ欠損 |
| OCA2 | OCA2(P遺伝子) | メラノソームpH調節1) |
| OCA3 | TYRP1 | ユーメラニン合成 |
| OCA4 | SLC45A2 | メラノソーム輸送体 |
| OCA8 | DCT(TYRP2) | ドパクロム変換2) |
| OA1 | GPR143 | メラノソームシグナル3) |
| HPS1/HPS4 | HPS1/HPS4 | BLOC-3複合体4) |
| HPS11 | BLOC1S5 | BLOC-1複合体5) |
OCA2タンパク質の機能:12膜貫通αヘリックス構造を持つNa⁺/H⁺アンチポーターファミリーに属する1)。メラノソーム特異的アニオンチャネルの構成要素として、ステージI/IIメラノソームのpHを塩素電流制御によって調節する1)。ClinVarには477の病原性変異が登録されている1)。
DCT(TYRP2):ドパクロムからDHICA(5,6-ジヒドロキシインドール-2-カルボン酸)への変換を触媒する2)。OCA8の原因遺伝子である。
GPR143(OA1):メラノソームの小胞輸送を制御するシグナル伝達分子。変異によりマクロメラノソームが形成される3)。
HPS(BLOC複合体):HPS1/HPS4はBLOC-3複合体としてRab32/38のGEF(グアニンヌクレオチド交換因子)として機能する4)。BLOC-1は8サブユニットからなりエンドソームリサイクリングに関与し、BLOC1S5(BLOS3)遺伝子変異がHPS-11(2020年初記載)を引き起こす5)。
タイプによって視力予後・全身合併症の有無・遺伝形式が異なるため、正確なサブタイプ診断が重要である。特にHPSは出血傾向・肺線維症・炎症性腸疾患を合併し、手術や抜歯前の注意が必要である。遺伝子検査によるサブタイプ同定は適切な管理計画の立案に直結する。
典型的なOCAは白髪・白皮膚・眼所見(虹彩透照・眼振)の組み合わせから臨床的に診断できる。ただし日本人OA1では虹彩に色素が残存するため、眼症状のみで受診する場合は診断が遅れることがある。
根治的治療は存在しない。治療は視機能の最大化と合併症の管理が目標となる。
視覚管理
屈折矯正:乳幼児期からの眼鏡矯正が基本。弱視予防を目的とした早期介入が重要。
遮光眼鏡:室内では約20%遮光、屋外では約80%遮光のレンズを使用する5)。
虹彩付きコンタクトレンズ:美容的改善と羞明軽減を目的として使用できる。
ロービジョンケア:拡大鏡・拡大読書器・タブレット端末等の補助器具を活用する。
眼振・斜視
斜視手術:整容目的で施行される。眼振の軽減を期待したDawson-Trick-Litzkow(DTL)手術が行われる場合もある。
眼振治療:根治的治療はない。異常頭位(null zone確保)への対応として斜視手術が検討される。
皮膚・全身管理
日光防御:UVB遮断の日焼け止め・衣服・帽子による皮膚保護。日本人でも扁平上皮がんリスクに注意。
HPS管理:NSAIDs(アスピリン含む)は血小板機能異常を悪化させるため原則禁忌。10代からの定期的な肺機能モニタリングが推奨される4)。
遺伝カウンセリング:家族計画・保因者診断・サブタイプ確定のために必須。
HPSに合併する肺線維症に対しては、抗線維化薬が用いられる。
Liuら(2025)は新規ホモ接合HPS4変異を持つHPS症例で、ニンテダニブ投与により肺線維症が18ヵ月間安定化したことを報告した4)。ピルフェニドンも同様に用いられる治療選択肢である。
現時点で白皮症に対する遺伝子治療の実用化はされていない。レチノスキシスやLCAなど他の遺伝性網膜疾患では遺伝子治療が承認されており(例:Luxturna)、白皮症への応用研究が進んでいる6)。現在は屈折矯正とロービジョンケアが標準治療の中心である。
メラニン合成はメラノソーム内で以下の経路をたどる。
チロシン → L-DOPA → DOPAキノン → (ユーメラニンまたはフェオメラニン)
ドパクロムからDHICA(5,6-ジヒドロキシインドール-2-カルボン酸)への変換はDCT(TYRP2)が触媒し2)、ユーメラニン合成に必須の反応である。
OCA2のメラノソーム成熟障害:OCA2タンパク質の機能低下により、メラノソームはステージIV(成熟メラノソーム)への移行が障害され、ステージI/IIの未熟メラノソームが増加する1)。塩素電流制御によるpH調節の破綻が主たる機序である1)。
網膜色素上皮の色素沈着が不適切な場合、発達過程における光遮蔽作用が失われ、中心窩の形態的発達(avascular zone形成・錐体細胞の密集移動)が阻害される。これが白皮症における視力不良の根本原因となる。
白皮症では耳側網膜からの線維が視交叉で過剰に対側に交叉し、通常よりも多くの線維が鼻側として処理される。この「misrouting」は視交叉係数の非対称化としてVEPで捉えられる2)。
Rateauxら(2025)のOCA8研究では、DCT変異によりL-DOPAが野生型の50%に低下するマウスモデルを用い、L-DOPA補充により眼科的異常が回復することが示された2)。L-DOPAがメラニン合成中間体として視路形成に果たす役割が示唆されている。
GPR143変異(OA1):GPR143タンパク質の機能喪失により、メラノソームの小胞輸送が障害されてマクロメラノソームが形成される3)。
HPS(BLOC複合体障害):BLOC-3の機能喪失によりRab32/38の活性化が障害され、メラノソーム関連細胞小器官(LRO: lysosome-related organelle)の生合成が全般的に破綻する4)。血小板内のdense bodyや肺II型上皮細胞内ラメラ小体が影響を受ける。肺線維症の背景には、異常なラメラ小体からのセロイド様物質蓄積が関与するとされる4)。
BLOC-1(HPS-11):BLOC1S5(BLOS3)を含む8サブユニット複合体として機能し、エンドソームリサイクリングを制御する5)。
L-DOPAはメラニン合成の中間体であるとともに、網膜発達・視路形成に重要な役割を果たす。
Rateauxら(2025)のOCA8マウスモデルでは、DCT変異によるL-DOPA低下が眼科的表現型の主要因であり、L-DOPA補充により異常が改善された2)。ヒトへの応用が研究されている6)。
白皮症患者を対象としたレボドパ投与による視機能改善の評価が進行中であり、網膜発達促進効果が期待されている6)。
4-ヒドロキシフェニルピルビン酸ジオキシゲナーゼ(4-HPPD)阻害薬であるニチシノンは、チロシン代謝経路を調節することで眼内メラニンを増加させる可能性が研究されている6)。
OCA関連遺伝子(TYR・OCA2等)を標的とした遺伝子補充療法の基礎研究が進んでいる。LCA(レーバー先天性黒内障)に対するLuxturna(RPE65遺伝子補充)の承認が、遺伝性網膜疾患全般の遺伝子治療研究を加速させている6)。
Jiangら(2024)は中国人家族の全エクソーム解析(WES)により、OCA2遺伝子の新規複合ヘテロ接合変異(c.635A>G/c.2359+1G>T)を同定した1)。変異タンパク質は野生型と比較してメラノサイトへの移行が障害されることが機能解析で確認された。
Boeckelmannら(2021)は、BLOC1S5変異によるHPS-11を2020年以降報告された症例を含む5例の検討で、本症候群の臨床スペクトラムを詳述した5)。肺・眼・神経症状のモニタリング指針が示されている。
HPS肺線維症に対するニンテダニブ・ピルフェニドンによる抗線維化療法の有効性が継続的に評価されている4)。
Flynnら(2025)はGPR143変異を有するOA1保因者女性の眼底検査で、94%に弓状脈前方の斑状色素変化、74%に虹彩透照が確認されたと報告した3)。保因者でも眼科的異常が高率に認められることが示された。
OCAでは皮膚色素が欠如するため、紫外線によるDNA損傷が蓄積しやすく、扁平上皮がんなどの皮膚がんリスクが有意に上昇する。アフリカ系OCA患者では特にリスクが高いとされる。定期的な皮膚科受診と徹底した日光防御(日焼け止め・遮蔽衣服)が推奨される。
Jiang B, Zhang H, Kan Y, Gao X, Du Z, Liu Q. Novel compound heterozygous mutations in OCA2 gene were identified in a Chinese family with oculocutaneous albinism. Mol Genet Genomic Med. 2024;12:e2297.
Rateaux M, Hadj-Rabia S, Barrois R, Zambrowski O, Michaud V, Moreno-Artero E, Bremond-Gignac D, Javerzat S, Robert MP. Chiasmal decussation in oculo-cutaneous albinism type 8. Invest Ophthalmol Vis Sci. 2025;66(2):44.
Flynn E, Cheela I, Kaden TR. Female carrier of ocular albinism linked to Gpr143 gene. J VitreoRetinal Dis. 2025;1-4.
Liu Q, Qing W, Guo S, Wang Y, Chen Y, Liu J. A novel homozygous HPS4 mutation in Hermansky-Pudlak syndrome: case report and literature review. Ther Adv Respir Dis. 2025;19:1-8.
Boeckelmann D, Wolter M, Kasmann-Kellner B, Koehler U, Schieber-Nakamura L, Zieger B. A novel likely pathogenic variant in the BLOC1S5 gene associated with Hermansky-Pudlak syndrome type 11 and an overview of human BLOC-1 deficiencies. Cells. 2021;10:2630.
Gurnani B, et al. Nystagmus review. Clin Ophthalmol. 2025;19:1617-1648.