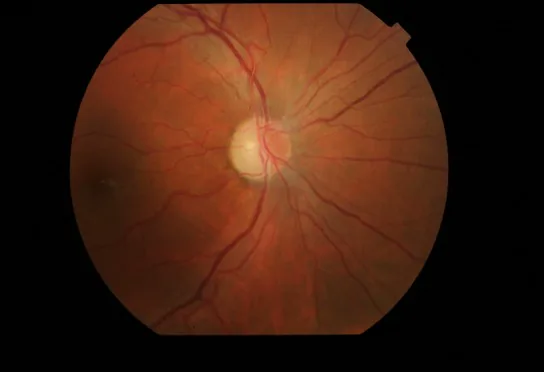
急性期
視神経乳頭の発赤・充血:乳頭周囲のRNFL浮腫を伴う。
網膜毛細血管拡張:毛細血管の拡張(telangiectasia)と血管蛇行の増加を認める。
蛍光眼底造影での非漏出:発赤した視神経乳頭から蛍光色素の漏出を認めない。これが視神経炎との重要な鑑別点である。
OCT所見:発症前より視神経乳頭の腫脹所見を認めるとされる。

Harding病は、レーバー遺伝性視神経症(Leber hereditary optic neuropathy; LHON)のミトコンドリアDNA(mtDNA)変異を背景に、多発性硬化症(MS)に類似した脱髄性神経症状が共存する疾患である。1992年にHardingらが、LHON家族歴を持つ両眼性視神経症の女性8例(うち6例にMSに合致する神経症状)を報告したことに由来する。1)
文献上これまでに計88例が報告されている。1)患者の70.4%(62例)が女性であり、男女比は2.38:1である。1)LHONが男性に多い(男性93.1%)ことと対照的であり、MSの女性優位な素因が関与すると考えられる。平均発症年齢は30.5歳である。1)
LHONの有病率は日本で約1/50,000(国内総患者数約4,000〜5,000人と推計)、英国で1/31,000、オーストラリアで1/68,000とされる。1)2015年に日本でLHONは難病指定され、診断基準が確立された。新規発症者数は年間約117人で、30歳代までの発症が47%を占める。
文献上の報告症例は88例にとどまり、きわめてまれな疾患である。1)患者の70.4%が女性で平均発症年齢は30.5歳である。LHONとMSという2つの疾患の共存が必要であるため、単独のLHONやMSよりはるかに少ない。
急性期
視神経乳頭の発赤・充血:乳頭周囲のRNFL浮腫を伴う。
網膜毛細血管拡張:毛細血管の拡張(telangiectasia)と血管蛇行の増加を認める。
蛍光眼底造影での非漏出:発赤した視神経乳頭から蛍光色素の漏出を認めない。これが視神経炎との重要な鑑別点である。
OCT所見:発症前より視神経乳頭の腫脹所見を認めるとされる。
慢性期
視神経乳頭萎縮:炎症収束後に乳頭萎縮が残存する。
RNFL菲薄化:OCTで乳頭黄斑線維束を主体とした網膜内層の菲薄化が認められる。
最終矯正視力の低下:0.01前後にとどまることが多い。光覚は保たれる。
対光反射の保存:他の視神経疾患に比べ対光反射が保たれるか、障害されても軽微である。
LHONとの違いは、複数回の視力低下エピソードが生じること、2眼目が侵されるまでの間隔が長いこと(平均1.66年)である。1)MSの視神経炎との違いは、眼痛を伴わないこと、蛍光眼底造影で視神経乳頭からの色素漏出を認めないことである。これらの特徴がHarding病の鑑別に重要な手がかりとなる。
Harding病の原因はミトコンドリアDNA(mtDNA)の病原性変異である。主要3変異はいずれも複合体Iサブユニットをコードし、ATP合成障害と活性酸素(ROS)増加を介して網膜神経節細胞(RGC)のアポトーシスを誘導する。3)
Harding病88例における変異の内訳を以下に示す。
| 変異 | 遺伝子 | Harding病での割合 |
|---|---|---|
| m.11778G>A | MT-ND4 | 69.3%(61/88例) |
| m.14484T>C | MT-ND6 | 12.5% |
| m.3460G>A | MT-ND1 | 10.2% |
引用: Alorainy J et al. 20241)
MT-ND4変異はアジアではLHON全体の90%、欧州では70%を占める。3)国内患者でもこの3遺伝子変異が95%を占める。
mtDNAは母親から子に伝わる(母系遺伝)。男性患者の子孫には遺伝しない。
LHONとMSが共存する機序として、以下の3つの仮説が提唱されている。1)
LHONは男性優位(男性93.1%)であるが、Harding病は患者の70.4%が女性である。これはMSが女性に多い疾患であることと関連する。MSの素因となる遺伝的・環境的要因が、無症状のmtDNA変異保持者にLHONを発症させるという仮説が提唱されている。1)またエストロゲンシグナル伝達がLHONの病態に関与する可能性も示唆されている。2)
Harding病は「MS診断基準を満たし、かつ主要なLHON変異を有する患者」と定義される(Pfefferら)。
日本では2015年に確立されたLHONの診断基準が適用される。
| 検査 | 主な目的・所見 |
|---|---|
| mtDNA解析・マルチジーンパネル | m.3460G>A・m.11778G>A・m.14484T>Cの特定 |
| MRI | 脱髄性T2高信号白質病変の確認 |
| 蛍光眼底造影 | 視神経乳頭からの色素漏出の有無(非漏出が鑑別点) |
| 光干渉断層計(OCT) | 乳頭黄斑線維束の菲薄化 |
| 心電図(ECG) | 心臓早期興奮症候群の除外 |
中毒性視神経症、常染色体優性視神経萎縮(ADOA)、オカルト黄斑ジストロフィ、錐体ジストロフィ、圧迫性視神経症、視神経脊髄炎スペクトラム(NMOSD)との鑑別が必要である。
確立した治療法はない。以下に現状の対応を示す。
標準治療の確立していない中で、以下の治療が試みられている。
確立した治療法はなく、対症療法が中心である。国内未承認のイデベノンを個人輸入して内服する患者もいるが、Harding病での有効性エビデンスは乏しい。4)CoQ10やビタミンB群などのサプリメント、禁煙指導、ロービジョンケア、遺伝カウンセリングが行われるのが現状である。
LHONの基本的機序は、mtDNA変異による複合体I(ミトコンドリア呼吸鎖)サブユニットの障害に端を発する。電子伝達の障害によりATP合成が低下し、同時に活性酸素(ROS)が蓄積する。これがRGCのアポトーシスを誘導し、視神経萎縮に至る。3)
変異別の視力回復率には差があり、MT-ND4変異では視力回復率が4〜25%とMT-ND1・MT-ND6変異より低い。3)国内で最も多いmt11778(MT-ND4)の視機能改善率は数パーセント程度にとどまる。mt14484(MT-ND6)変異が最も改善率が高い。
LHONの浸透率が男性に高いことから、X染色体連鎖の核遺伝子の関与が示唆されている。3)
LHON-MSの組織病理では、T細胞と活性化マクロファージ/ミクログリアが組織障害を媒介する。LHON病変内に炎症細胞が存在することは通常と異なり、初期の免疫学的機序を示唆する。1)白質変化はMS様の脱髄だけでなく、空胞化やびまん性髄鞘蒼白も寄与する。ミトコンドリア機能障害・自己免疫応答・分子擬態が炎症性脱髄の機序として推定される。1)
神経学的後遺症は軽度の障害から再発寛解型MSに似た経過まで多岐にわたる。
MT-ND4変異のLHON患者を対象とした遺伝子治療(レナドゲン・ノルパルボベック; rAAV2/2-ND4)が開発されている。アロトピック遺伝子発現により野生型ND4タンパク質をRGCのミトコンドリアに導入する。2)
REVERSE study(2019)では、硝子体内注射眼で平均BCVA改善 -0.308 LogMAR、未処置眼で -0.259 LogMARが報告された。2)対側眼への免疫学的移行が示唆される結果であった。プール解析(RESCUE, REVERSE, RESTORE, REFLECTの4試験)では対照群比で約21.5 ETDRS文字の改善が示された。両側投与が片側投与より効果が大きく(約12 vs 約8 ETDRS文字)、メタ解析でもrAAV2/2-ND4はイデベノンより有効、いずれも自然経過より有効であった。2)
現在EMAおよびFDA未承認であり、Harding病に対する有効性は未検証である。LHONコンポーネントへの効果が期待される一方、MS病態への影響は明らかでない。2)
iPSC由来MSCを硝子体内投与し、RGCにミトコンドリアを直接移行させる手法が研究されている。F-actin依存性のトンネリングナノチューブ(TNT)が移行を仲介する。Ndufs4 KOマウスにおいてRGC密度低下の防止が報告されているが、臨床試験には至っていない。3)
MSにおける視覚誘発電位(VEP)改善の可能性が示唆されており、視覚予後不良なHarding病患者への応用が検討されている。
Harding病に特化したバイオマーカーの開発と前向き研究が必要とされる。LHONとMSの二重の病態を持つ本疾患に対する治療は個別化されるべきであり、多職種連携が不可欠である。
rAAV2/2-ND4はMT-ND4変異のLHON患者を対象とした遺伝子治療であり、LHONコンポーネントへの効果が期待される。2)ただしHarding病に対する有効性は未検証であり、現在EMAおよびFDA未承認である。MS病態への影響も明らかでなく、本疾患への応用には慎重な検討が必要である。