MEN1型
責任遺伝子:MEN1遺伝子(11q13)
腫瘍の組み合わせ:副甲状腺腫瘍、下垂体腫瘍、胃腸膵管腫瘍
有病率:10万人あたり2〜20人
眼科的特徴:下垂体腫瘍による視交叉圧迫(両耳側半盲)
多発性内分泌腫瘍症(MEN)は、2つ以上の内分泌腺において新生物が発生するリスクが高まる稀な遺伝性疾患群である。常染色体優性遺伝の形式をとり、高い浸透率と多様な表現度を示す。
MENは主に4つの型に分類される。
MEN1型
責任遺伝子:MEN1遺伝子(11q13)
腫瘍の組み合わせ:副甲状腺腫瘍、下垂体腫瘍、胃腸膵管腫瘍
有病率:10万人あたり2〜20人
眼科的特徴:下垂体腫瘍による視交叉圧迫(両耳側半盲)
MEN2型(2A・2B)
責任遺伝子:RET原癌遺伝子(10q11.2)
腫瘍の組み合わせ:甲状腺髄様癌(MTC)、褐色細胞腫、副甲状腺機能亢進症(2Aのみ)
有病率:35,000人に1人(MEN2型全体)
眼科的特徴:著明な角膜神経肥厚、結膜・眼瞼神経腫(特にMEN2B型)
MENの眼科的特徴は、古典的にはMEN2B型において記述されている。MEN2B型では甲状腺髄様癌、褐色細胞腫に加え、粘膜神経腫と腸管神経節細胞腫症を合併する。眼科的所見がMEN2B型の早期診断の手がかりとなることがある。
MEN1型では下垂体腫瘍(患者の20〜30%に発生)が視交叉を圧迫し、視野欠損を引き起こす可能性がある。プロラクチノーマが最も一般的な下垂体腫瘍である。
MEN2B型の特徴的所見は小児期から認識可能である。角膜神経の肥厚や粘膜神経腫は早期に出現し、MEN2B型の最初の臨床的手がかりとなることがある。MEN2B型のMTCは乳児期に発症することもあり、眼科的所見の早期認識が診断と予防的治療の時期を左右する。
MEN2B型とMEN1型では眼科的な訴えが異なる。
| MEN2B型の眼科的所見 | 特徴 |
|---|---|
| 著明な角膜神経 | 最も一般的な所見 |
| 結膜・眼瞼神経腫 | 被膜のない神経増殖 |
| ドライアイ | 涙液分泌異常 |
| 眼瞼肥厚 | 神経腫による |
| 開放隅角緑内障 | 稀 |
著明な角膜神経はMEN2B型における最も特徴的な眼科的所見である。細隙灯顕微鏡検査で角膜内の肥厚した神経線維を確認する。組織病理学的にはシュワン細胞を伴う無髄神経であり、軸索は正常な外観を示す。
結膜・眼瞼の神経腫は、被膜のない肥厚した神経増殖として認められる。口唇、舌、頬粘膜にも同様の粘膜神経腫が出現する。
MEN1型では下垂体腫瘍による視交叉症候群が主な眼科的問題である。視野検査で垂直経線を境界とする両耳側半盲が最も高頻度に認められる。視神経萎縮を伴うこともある。
**非眼科的特徴(MEN2B型)**として、口唇肥大、マルファン症候群様体型、腸管神経節細胞腫症(巨大結腸症を引き起こしうる)が認められる。
MEN症候群は常染色体優性遺伝の形式をとる。RET変異を持つ個人では子孫への遺伝リスクが50%である。体細胞変異による孤発例も存在する。
MEN1遺伝子(染色体11q13)の変異が原因である。MEN1遺伝子はメニン(Menin)というタンパク質をコードする癌抑制遺伝子である。両方のアレルに変異が生じる「2ヒット」により機能が喪失し、制御不能な細胞増殖が起こる。
RET原癌遺伝子(染色体10q11.2)のミスセンス変異が原因である。RETタンパク質はチロシンキナーゼ受容体であり、神経内分泌組織の分化と移動に関与する。変異により機能獲得型の異常が生じ、腫瘍が発生する。
MEN2B型では95%の患者にコドン918(M918T変異)の変異が認められる。この変異を持つ患者は乳児期から発症し、最も侵襲性の高いMTCを呈する。
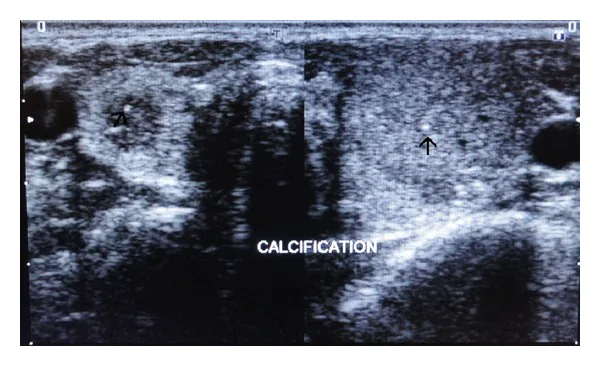
MEN症候群の診断は、通常、家族歴と非眼科的所見に基づいて行われる。眼科検査は特に新規症例での精査に有用である。
著明な角膜神経が観察される疾患として以下が挙げられる。
| 鑑別疾患 | 特徴 |
|---|---|
| レフサム症候群 | フィタン酸蓄積 |
| ハンセン病 | 末梢神経障害 |
| ライリー・デイ症候群 | 家族性自律神経失調 |
| 神経線維腫症 | カフェオレ斑 |
| 先天性魚鱗癬 | 皮膚角化異常 |
純粋粘膜神経腫症候群(MNS)の患者はMEN2B型と同様の眼科的特徴を呈することがある。
MEN患者の管理には多職種連携が不可欠である。臨床遺伝専門医、内分泌専門医、外科医、腫瘍専門医が関与する。
遺伝カウンセリングはRET遺伝子検査に先立って行う。MEN2型の家系では出生時または可能な限り早期にスクリーニングを実施する。新たにRET変異が診断された初発例の第一度近親者全員に遺伝カウンセリングを提供すべきである。
MEN2B型のコドン883、918、922の生殖細胞系列変異を持つ患者では、生後数ヶ月以内に甲状腺全摘除術と中央区域リンパ節郭清を行う。MTCの生涯浸透率がほぼ100%であるためである。
眼科医はMENにおける眼科的発現の早期発見、適切な紹介、視能リハビリテーションに重要な役割を担う。MEN1型の下垂体腫瘍では定期的な視野検査と視力評価が必要である。MEN2B型ではドライアイの管理と定期的な眼圧測定を行う。
MEN2B型では定期的な細隙灯顕微鏡検査で角膜神経や結膜・眼瞼神経腫の変化を観察する。ドライアイ症状の有無を確認し、必要に応じて人工涙液などで管理する。開放隅角緑内障のリスクがあるため、定期的な眼圧測定と視野検査も推奨される。また、下垂体腫瘍の合併がないかについても画像検査で評価する。
MEN1型は「2ヒット仮説」に基づく癌抑制遺伝子の機能喪失で発症する。第1の変異(生殖細胞系列変異)は遺伝により受け継がれ、第2の変異(体細胞変異)が後天的に加わることでメニンタンパク質の機能が完全に喪失し、細胞増殖の制御が失われる。
メニンは内分泌組織と非内分泌組織の両方で発現するが、発癌に至る正確な機序は未解明である。下垂体腫瘍が視交叉を圧迫することで両耳側半盲を生じる。
RETタンパク質は発達中の神経内分泌組織の分化と移動に関与するチロシンキナーゼ受容体である。RET遺伝子のミスセンス変異は単一アミノ酸置換をもたらし、タンパク質の恒常的活性化(機能獲得)を引き起こす。
RETは甲状腺傍濾胞細胞(C細胞)、副甲状腺、腸管神経節、副腎クロム親和性細胞、末梢・中枢神経系を含む複数の組織で発現している。この広範な発現パターンがMEN2型における多様な腫瘍パターンを説明する。
MEN2B型における肥厚した角膜神経の組織病理学的検査では、シュワン細胞を伴う無髄神経が示される。軸索は正常な外観を呈し、直径は0.1〜1.4nmの間で変化する。結膜・眼瞼の神経腫は被膜を持たない神経増殖として認められる。