母斑症(神経皮膚症候群)は皮膚・神経系・眼に過誤腫性病変を生じる先天性疾患群の総称である。
代表疾患はNF 1・NF2・結節性硬化症・スタージ・ウェーバー症候群・フォン・ヒッペル・リンドウ病・毛細血管拡張性運動失調症の6疾患である。
NF1ではLisch結節(虹彩 過誤腫)が成人の90%以上に認められ、診断上重要な眼所見である。
SWSでは30〜70%に緑内障 が発生し、先天緑内障では手術が必須となる。
VHLでは網膜毛細血管腫 が患者の約60%に生じ、1歳から年1回の散瞳 眼底検査 が推奨される。
各疾患の眼合併症は無症状のうちに進行することが多く、定期的な眼科的管理が不可欠である。
分子標的薬(MEK阻害薬、mTOR阻害薬など)が一部疾患で標準治療または新規治療として導入されている。
母斑症(phakomatoses)は、皮膚・中枢神経系・眼に過誤腫性病変を特徴とする先天性疾患群の総称である。神経皮膚症候群(neurocutaneous syndromes)とも呼ばれる。
命名はオランダの眼科医Van der Hoeve(ヴァン・デル・フーフェ)によるもので、ギリシャ語の「phakos(レンズ・斑点)」に由来する。当初は神経線維腫症・結節性硬化症・フォン・ヒッペル・リンドウ病の3疾患が含まれ、その後スタージ・ウェーバー症候群・毛細血管拡張性運動失調症が追加された。現在は60以上の症候群が記載されている。
共通する病態基盤は、神経堤細胞の形成・移動・分化の異常である。神経堤細胞は外胚葉由来でシュワン細胞・メラノサイトなど多様な細胞を産生するため、神経・皮膚・眼の多臓器に病変が生じる。関与するシグナル経路としてRAS・MAPK/MEK・mTOR・PI3K/AKT・GNAQ・VHL-HIF経路が知られている。
主要6疾患の発生頻度は以下の通りである。
疾患 発生頻度(人に1人) NF1(神経線維腫症1型) 3,000〜5,000 結節性硬化症(TSC ) 6,000〜10,000 SWS(スタージ・ウェーバー症候群) 20,000〜50,000 VHL(フォン・ヒッペル・リンドウ病) 36,000 NF2(神経線維腫症2型) 25,000〜100,000 AT(毛細血管拡張性運動失調症) 88,000〜100,000未満
Q 母斑症にはどのような疾患が含まれるか?
A 代表的な6疾患はNF1・NF2・結節性硬化症・スタージ・ウェーバー症候群・フォン・ヒッペル・リンドウ病・毛細血管拡張性運動失調症である。いずれも遺伝子変異に起因し、神経・皮膚・眼の多臓器に病変を生じる。現在は60以上の症候群が母斑症の範疇に含まれる。
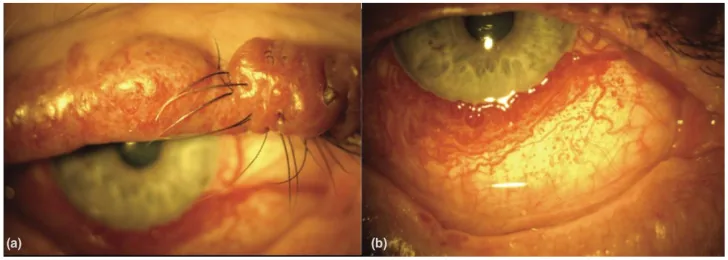
phakomatoses sturge weber eyelid conjunctival photo An Update on Multimodal Ophthalmological Imaging of Diffuse Choroidal Hemangioma in Sturge–Weber Syndrome. Vision (Basel). 2023 Oct 6; 7(4):64. Figure 1. PMCID: PMC10594527. License: CC BY.
(a) Port-wine stain of the upper lid with nodularity in a patient with Sturge–Weber syndrome. (b) Diffuse conjunctival vascularity in a patient with Sturge–Weber syndrome. From [15].
各疾患で眼合併症の種類が異なり、自覚症状も多様である。
NF1 :視路腫瘍(視神経 膠腫)が進行すると、視力 低下・色覚喪失・視野欠損 が生じる。蔓状神経線維腫では眼球突出 をきたすことがある。結節性硬化症 :網膜星細胞過誤腫 は通常無症状である。黄斑 や視神経乳頭を侵した場合に視力障害をきたす。SWS :先天緑内障を伴う場合、角膜 混濁・流涙・羞明 が生じる。後期発症の緑内障では無痛性の進行性視野欠損が起こる。VHL :早期は無症状。網膜毛細血管腫が進行すると滲出性変化・黄斑浮腫 ・輪状白斑が出現し、視力低下をきたす。AT :視力は通常維持される。眼球運動障害 (眼振 ・眼球運動失行 )が主体である。
Lisch結節(リッシュ結節) :NF1で最も頻度の高い眼所見。淡褐色・境界鮮明・ドーム状の小結節(1〜2mm未満)が虹彩に多発する。年齢別有病率は3歳未満5%・3〜4歳42%・5〜6歳55%・21歳以上100%と年齢とともに増加し、NF1の診断基準(2個以上)に含まれる。日本人では虹彩色が茶系のため、細隙灯による精査が重要である。視路腫瘍(視神経膠腫) :NF1患者の約15〜25%に合併。低悪性度毛様細胞性星細胞腫が多く、無症候性のことが多い。進行すると視神経萎縮 ・視力障害・視野障害をきたす。視交叉 浸潤例もある。蔓状神経線維腫 :NF1患者の10%未満に発生。眼瞼の「S字型変形」が特徴で、触診で「虫袋(bag of worms)」様の感触がある。眼球突出・斜視 ・弱視 ・先天緑内障の原因となる。蝶形骨翼形成不全 :眼窩 骨壁の先天性欠損で、拍動性眼球突出を生じることがある。緑内障 :NF1の1〜2%に発生。先天性(片眼性)と遅発症の2タイプがある。その他 :角膜有髄神経の著明化、脈絡膜 肥厚、網膜 有髄神経。
視神経鞘髄膜腫 白内障 網膜・RPE 過誤腫 、網膜上膜 :合併することがある。露出性角膜炎 :両側聴神経腫瘍による第5・第7脳神経障害が顔面しびれ・複視 ・兎眼 を引き起こし、続発する。Lisch結節はまれである。
網膜星細胞過誤腫 :約50%に合併。3型に分類される。(i)平坦・半透明・非石灰化型、(ii)隆起・多結節・石灰化型(「桑の実様(mulberry)」外観)、(iii)移行型。通常は後極に複数個みられる。網膜脱色素病変 :「打ち抜き状(punched-out)」外観を呈する。網膜血管異常 :動脈瘤様拡張・動静脈奇形を生じ、硝子体出血 ・増殖性硝子体網膜症 ・網膜剥離 の原因となりうる。その他 :眼瞼血管線維腫、虹彩脱色素斑、非定型的脈絡膜欠損。
三主徴は(1)三叉神経 領域の顔面血管腫、(2)同側頭蓋内血管腫、(3)同側の緑内障または脈絡膜血管腫である。
緑内障 :SWSで最も重要な眼所見。30〜70%に発生。先天緑内障(生後〜4歳)が約60%を占め、牛眼・角膜混濁・巨大角膜 をきたす。病因は隅角 発育異常・上強膜 静脈圧上昇・脈絡膜血管腫の関与が考えられている。眼瞼血管腫の存在下で高頻度に発生する。脈絡膜血管腫 :約20〜70%に発生。びまん性で境界不鮮明なため通常の眼底検査では同定が困難である。眼底は「トマトケチャップ状」外観を呈する。通常は増大傾向を示さないが、滲出性変化・滲出性網膜剥離 をきたすことがある。その他 :結膜 ・上強膜・虹彩の血管拡張蛇行。
網膜毛細血管腫(血管芽腫) :VHL患者の43〜85%(国内報告では約60%)に発生。約1/3が両眼性で多発性。網膜耳側周辺部に好発し、拡張蛇行した輸出入血管を伴う赤橙色結節として観察される。平均発症年齢は25歳で、通常30歳までに出現する。進行すると滲出性変化・網膜出血→黄斑浮腫・輪状白斑→視力低下をきたす。
さらに進行すると硝子体出血・牽引性網膜剥離 ・血管新生緑内障→失明に至りうる。
蛍光眼底造影 では輸入動脈への色素流入→輸出静脈灌流の所見を示し、腫瘤部で早期より著明な色素漏出がみられる。
球結膜毛細血管拡張 :最も一般的な眼所見。通常5〜8歳までに発生し、患者の80〜90%にみられる1) 。眼球運動障害 :眼振・眼球運動失行(oculomotor apraxia)・サッケード 異常・輻輳と調節の異常・斜視が認められる。視力は通常維持される。
Q Lisch結節は年齢によってどのように変化するか?
A NF1に伴うLisch結節は年齢とともに出現頻度が上昇する。3歳未満では5%にとどまるが、21歳以上ではほぼ100%に認められる。若年では細隙灯検査でも検出困難な場合があり、年齢を考慮した解釈が必要である。
各疾患の遺伝形式・原因遺伝子・染色体座位を以下に示す。
疾患 遺伝形式 原因遺伝子 染色体座位 NF1 常染色体優性 NF1 17q11.2 NF2 常染色体優性 NF2 22q11.1-q13.1 TSC 常染色体優性 TSC1/TSC2 9q34/16p13 VHL 常染色体優性 VHL 3p25-26 SWS 孤発性(体細胞モザイク) GNAQ 9q21 AT 常染色体劣性 ATM 11q22
各疾患の遺伝的特徴は以下の通りである。
NF1 :約50%がde novo変異。ほぼ完全浸透率だが表現型は多様。ニューロフィブロミンはRAS-GAPとして機能し、GTP-RASをGDP-RASに変換する2) 。NF2 :マーリン(merlin)タンパク質の機能喪失。シュワン細胞・軟膜細胞に主に発現する腫瘍抑制因子で、欠損するとシュワン腫・髄膜腫・上衣腫が生じる。TSC :散発例が約2/3を占める。TSC2変異は散発例の75〜80%を占め、TSC1変異より重症の表現型をとる1) 。ハマルチン/ツベリンはmTOR経路を直接阻害する2) 。SWS :孤発性でGNAQ遺伝子の体細胞モザイク変異が原因1) 。三叉神経第1枝(V1)領域全体のポートワイン母斑(PWB)は眼科・神経学的合併症のリスクが高い。VHL :浸透率はほぼ100%で、約20%がde novo変異。pVHLはHIF-αのユビキチン化・プロテアソーム分解に関与する2) 。Type 1(褐色細胞腫なし)が約80%、Type 2(褐色細胞腫あり)が約20%2) 。AT :常染色体劣性遺伝 でATMキナーゼはDNA二重鎖切断修復・ゲノム安定性維持に関与する1) 。悪性腫瘍(リンパ系)リスクおよび放射線感受性が著しく高い。
NF1の診断基準(NIH基準・日本皮膚科学会2008年)では、以下7項目のうち2つ以上を満たすことで確定診断される。
6個以上のカフェオレ斑(思春期前:最大径5mm以上、思春期後:15mm以上)
2個以上の神経線維腫またはびまん性神経線維腫
腋窩・鼠径部の雀卵斑様色素斑
視神経グリオーマ
2個以上のLisch虹彩結節
特徴的骨病変(蝶形骨異形成など)
第一度近親者に同症
眼科的検査の役割は以下の通りである。
細隙灯顕微鏡 :Lisch結節の検出(若年では困難なことがある)。前眼部OCT ・超音波生体顕微鏡 MRI :視路腫瘍の最良の診断法。視交叉・視索の評価に必須。視野検査 光干渉断層計 (OCT)網膜神経線維層 菲薄化の評価。VEP (視覚誘発電位)定期検査間隔 :Lisch結節のみ→年1回。視神経膠腫を有する場合→3か月に1回。
臨床基準では両側前庭神経シュワン腫が特徴的所見である。皮膚症状が乏しいためNF1より診断困難な場合がある。遺伝子検査では90%以上の変異が検出される。CT・MRIで両側聴神経腫瘍を確認する。
主要所見2つ、または主要所見1つ+副所見2つ以上で確定診断される。TSC1/TSC2病原性変異の同定は独立した診断基準として認められている1) 。
網膜星細胞過誤腫の評価には眼底検査・蛍光眼底造影・OCTを用いる。鑑別診断で最も重要なのは網膜芽細胞腫 である。小児期に石灰化を伴わない・栄養血管が乏しい・全身症状を伴うという特徴から鑑別が可能だが、桑の実状腫瘍との鑑別は困難なことがある。
Roach診断スケールでは、顔面ポートワイン母斑・眼圧上昇・軟膜血管腫のうち少なくとも2つを満たすことが基準とされる1) 。脈絡膜血管腫の評価にはED-OCT・MRIが有用である。頭部CTでは脳皮質内の石灰化が認められる。
緑内障のモニタリングには眼圧測定 ・視神経評価・視野検査を定期的に行う。乳幼児では細隙灯検査に加え、麻酔下での眼底検査・眼圧測定が必要となることがある。
遺伝子検査ではほぼ100%でVHL変異を検出でき、現在の確定診断の主流である。散瞳眼底検査は1歳から年1回の実施が推奨される。周辺部の赤橙色球状腫瘍+拡張蛇行血管が特徴的所見である。蛍光眼底造影は小さな周辺部血管芽腫の検出に有用で、OCTは小さな病変の評価に用いる。
臨床診断では運動失調・球結膜毛細血管拡張・眼球運動異常の三徴が重要である。血清AFP上昇・CA125上昇・ATMタンパク質欠損(ウェスタンブロット)が検査所見として認められる。球結膜毛細血管拡張は本疾患に特異的な所見(pathognomonic)である。MRIでは後頭蓋窩のびまん性小脳萎縮(特に虫部・半球)を示す1) 。
Lisch結節 :無症状の過誤腫であり治療は不要。視神経膠腫 :進行性の場合に外科的切除を検討するが、視機能は失われ術後合併症も多い。視交叉浸潤例では化学療法が適応となる。蔓状神経線維腫 :外科的全摘出は困難で再発しやすい。進行例では眼窩内容除去術 が必要になることがある。切除不能例にはセルメチニブ(MEK阻害薬)が新規治療として使用される1) 。緑内障 :通常の緑内障治療に準じた管理を行う。
前庭神経鞘腫に対しては外科的切除が基本で、3cm未満では65%で聴力温存が可能とされる1) 。露出性角膜炎は人工涙液・角膜保護眼鏡・必要に応じて瞼板 縫合で対処する。
網膜星細胞過誤腫 :通常は増大傾向を示さず、治療は不要である。網膜血管異常(動脈瘤様拡張・動静脈奇形) :硝子体出血・網膜剥離のリスクがあるため予防的光凝固を行う。硝子体出血・網膜剥離が発生した場合は硝子体手術 を検討する。てんかん :乳児の部分発作・点頭てんかんにはビガバトリンが第一選択とされる1) 。SEGA(上衣下巨細胞性星細胞腫)は神経外科的切除の適応となる1) 。
緑内障の治療 は発症時期によって方針が異なる。
先天緑内障(早期発症型) :手術治療が必須。線維柱帯切開術 ・隅角切開術が選択される。脈絡膜剥離・出血に対する十分な注意が必要であり、上強膜静脈圧上昇から通常より合併症リスクが高い。薬物治療への抵抗性が高い傾向がある。後期発症型 :まず薬物治療を行い、無効な場合は手術を検討する。
脈絡膜血管腫の治療 は滲出性変化の有無に応じて以下から選択する。
滲出性変化が生じた場合→光凝固
光凝固無効または滲出性網膜剥離→放射線治療(総線量20グレイ程度)。胞状網膜剥離の復位・血管腫縮小が期待できる。
難治例→硝子体手術
光線力学療法(PDT )・外照射放射線療法も選択肢となりうる3) 。
早期治療が原則である。
光凝固(第一選択) :アルゴンレーザーまたはダイレーザーを用い、血管腫周囲の網膜と栄養血管を密に凝固後、本体を直接凝固する。2乳頭径以上の腫瘍では流入動脈・周囲網膜を凝固後に腫瘍を直接凝固する。冷凍凝固 硝子体手術 :滲出性変化が強い場合・網膜剥離・増殖性変化が生じた場合に検討する。末期例 :血管新生緑内障に対する管理が必要となる。家族歴がある場合は家族全員の眼底検査を実施し、早期治療で予後良好が期待できる。
結膜毛細血管拡張に特異的な治療はない。免疫不全に対しては予防的抗菌薬・免疫グロブリン静脈内投与(IVIg )が行われる1) 。運動失調は対症療法が中心である。
Q VHLの網膜血管腫はいつから検査を受けるべきか?
A VHLでは1歳から年1回の散瞳眼底検査が推奨されている。VHL遺伝子変異が確認されている患者の家族も全員が眼底検査を受けることが重要で、早期治療により良好な視機能予後が期待できる。
各疾患の分子レベルの発症機序を以下に示す。
NF1・TSC(mTOR経路)
NF1(ニューロフィブロミン) :RAS-GAPとして機能。GTP結合型RAS(活性型)をGDP結合型RAS(不活性型)に変換促進。変異→RAS持続活性化→MAP kinase経路とPI3K-Akt-mTOR経路の脱制御→制御不能な細胞増殖2) 。
TSC(ハマルチン/ツベリン) :mTOR複合体1・2を直接阻害する腫瘍抑制複合体。変異→mTOR過活性→エネルギー代謝・タンパク/脂質合成・細胞生存の異常2) 。TSC2変異はTSC1変異よりも重症の表現型をとる1) 。
VHL(HIF経路)
pVHL :E3ユビキチンリガーゼ複合体(elongin B/C・Cullin 2・RBX1)の構成要素。正常酸素下ではプロリル水酸化酵素がHIF-αを水酸化→pVHLが認識→ユビキチン化→プロテアソーム分解。
VHL変異 :偽低酸素状態が恒常的に生じる→HIF-α蓄積→HIF-1βとのヘテロダイマー形成→VEGF・赤血球産生・代謝・細胞増殖関連遺伝子の転写活性化→血管腫瘍形成2) 。
NF2(マーリン) :腫瘍抑制タンパク質でシュワン細胞・軟膜細胞に発現。変異によりシュワン腫・髄膜腫・上衣腫が生じる。SWS(GNAQ体細胞モザイク変異) :Gタンパク質関連膜貫通シグナル伝達の異常。胚における変異時期・場所によって三叉神経領域・頭蓋内・眼における発現パターンが異なる1) 。GNAQはSWS・KTS・PPV に共通する分子基盤でもある3) 。AT(ATMキナーゼ) :DNA二重鎖切断修復・ゲノム安定性維持に関与する腫瘍抑制因子。変異→DNA修復障害→癌素因・放射線感受性の著しい増大・免疫不全1) 。
Chevalierら(2021)は母斑症と内分泌腫瘍の関連を検討し、NF1・TSC・VHLのシグナル経路(RAS-PI3K-Akt-mTOR経路、HIF経路)が多発性内分泌腫瘍の発生に共通して関与することを報告した2) 。
Q なぜ母斑症は多臓器に病変を生じるのか?
A 母斑症の共通病態基盤は、神経堤細胞の形成・移動・分化の異常である。神経堤細胞は外胚葉由来でシュワン細胞・メラノサイトなど多様な細胞を産生するため、神経・皮膚・眼の多臓器に病変が及ぶ。加えてRAS-mTOR経路やVHL-HIF経路など共通シグナル経路の変異が、臓器を超えた腫瘍発生を促進する。
切除不能な蔓状神経線維腫に対してFDA承認を受けた分子標的薬である。NF1のMAPK経路の過活性を標的とする1) 。
NF2関連シュワン腫・進行性腫瘍に対してINTUITT-NF2コンソーシアムでの臨床試験が実施されており、有望な結果が報告されている1) 。
VHL関連淡明細胞腎細胞癌・膵神経内分泌腫瘍を対象としたフェーズ2試験が進行中である。
Chevalierら(2021)によるMK6482の試験では、膵神経内分泌腫瘍の64%に客観的奏効が得られ、12か月無増悪生存率は98.3%と報告された2) 。
TSC関連腎血管筋脂肪腫・SEGAに対するEXIST試験で形態学的奏効が確認されており、内分泌腫瘍への応用も期待されている2) 。
GNAQ遺伝子変異がSWS・クリッペル・トレノネー症候群(KTS)・色素血管性母斑症(PPV)に共通の分子基盤であることが判明した3) 。これらの重複症例では脈絡膜メラノーマ のリスクがあるため、年1〜2回の散瞳眼底検査が推奨されている3) 。
Gama SM, Tamanini JVG, Moraes MPM, et al. A diagnostic approach to neurocutaneous syndromes. Arq Neuro-Psiquiatr. 2025;83(7):s00451809664.
Chevalier B, Dupuis H, Jannin A, et al. Phakomatoses and endocrine gland tumors: noteworthy and (not so) rare associations. Front Endocrinol. 2021;12:678869.
Senthilkumar VA, Kohli P, Mishra C, et al. Ocular features in a patient presenting with a rare combination of multiple phakomatoses. BMJ Case Rep. 2022;15:e252746.
記事の全文をコピーして、お好みのAIに貼り付けて質問できます
下のAIを開いて、チャット欄に貼り付け(ペースト) してください