第1期
傍中心窩RPE変化:網膜色素上皮(RPE)に点状・斑状の色素変化が現れる。
視力・自覚症状:ほぼ正常で経過することが多い。

中心性輪紋状脈絡膜ジストロフィ(Central Areolar Choroidal Dystrophy; CACD)は、黄斑部に境界明瞭な網脈絡膜萎縮を生じる遺伝性黄斑ジストロフィである。発症頻度は低く(10万人に1人程度)、希少疾患に分類される。
発症は20〜50歳代が多い。遺伝形式は常染色体優性遺伝(AD)が主体で、常染色体劣性遺伝(AR)や孤発例も報告されている。
最も頻度の高い原因遺伝子はPRPH2(染色体17p13)であり、ペリフェリン-2(peripherin-2)タンパクをコードする。ペリフェリン-2は視細胞外節ディスクの形成・安定化に不可欠なタンパクである。ハプロ不全(片方の対立遺伝子の機能喪失)が主要な発症機序と考えられている。
CACDにはABCA4遺伝子変異による疾患(スターガルト病など)との表現型の類似が指摘されており1)、遺伝子検査が鑑別に重要である。全身合併症は報告されていない。
加齢黄斑変性は通常60歳以降に発症し、ドルーゼンや滲出性変化を伴うことが多い。CACDは若年〜中年での発症が多く、境界明瞭な萎縮病変が特徴で、ドルーゼンは伴わない。遺伝子検査でPRPH2変異が確認されればCACDと診断できる。
両眼性の中心暗点(中心部が見えにくい)が主症状である。視力低下は早期から認められる場合もあるが、病期が進むまで比較的良好な視力が保たれることもある。
両眼に対称性の黄斑病変を呈し、病期は4段階に分類される。視神経・網膜血管・周辺部網膜は保たれる。萎縮域の大きさは概ね乳頭径(DD)の2〜4倍程度である。
第1期
傍中心窩RPE変化:網膜色素上皮(RPE)に点状・斑状の色素変化が現れる。
視力・自覚症状:ほぼ正常で経過することが多い。
第2期
境界不明瞭な低色素萎縮:中心窩を外れた部位に淡い萎縮域が出現する。
RPE変化の拡大:境界がまだはっきりしない萎縮が認められる。
第3期
境界明瞭なRPE萎縮:中心窩外に境界の明瞭な萎縮域が形成される。
中心窩温存:この段階では中心窩(fovea)が残存し、視力は比較的保たれる。
第4期
中心窩を含む完全萎縮:萎縮域が中心窩を含む黄斑全体に拡大する。
高度視力障害:脈絡毛細管板・視細胞・RPEの広範な消失により、視力が著しく低下する。
進行速度には個人差が大きい。一般に緩徐な経過をたどり、数十年かけて第4期へ至るケースもある。ただし遺伝子型によって進行速度が異なる場合があり、定期的な眼科的評価が不可欠である。詳細は「診断と検査方法」の項で述べるmfERGが早期進行の指標となる。
CACDの主因はPRPH2遺伝子の変異である。PRPH2はペリフェリン-2をコードし、視細胞外節ディスクの形成・安定化に機能する。変異はいずれも視細胞の正常な外節構造の維持を妨げる。
常染色体優性遺伝(AD)であるため、変異遺伝子を持つ親から子への遺伝確率は50%である。近親者の発症歴がある場合は、遺伝カウンセリングの受診が推奨される。
後天的なリスク要因や生活習慣による発症・進行促進因子は現時点では明確に同定されていない。
CACDは常染色体優性遺伝が主体であり、患者の子どもへの遺伝リスクは50%に及ぶ。遺伝子検査で変異を同定することで確定診断・家族スクリーニングが可能になる。また、将来の遺伝子治療の対象となる可能性があるため、変異の記録は重要である。
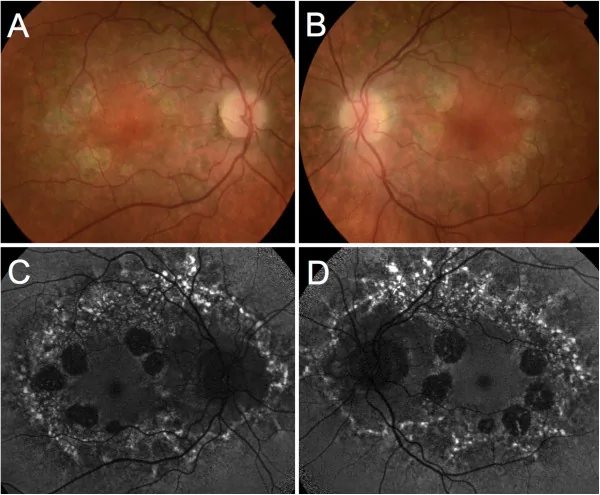
細隙灯顕微鏡・眼底鏡で黄斑部の境界明瞭な萎縮域を確認する。脈絡血管の露出や色素沈着の有無を評価する。
病期によって各検査の所見が異なる。下表に病期別の特徴的所見を示す。
| 検査 | 早期所見 | 進行期所見 |
|---|---|---|
| FAF(眼底自発蛍光) | 蛍光亢進 | 蛍光消失(萎縮域) |
| FA(蛍光造影) | 傍中心窩過蛍光 | 萎縮域の透見充填 |
| OCT | POS-RPE肥厚 | 外層(視細胞・RPE)消失 |
類似した黄斑萎縮を呈する疾患との鑑別が重要である。
| 鑑別疾患 | 鑑別のポイント |
|---|---|
| 萎縮型AMD | ドルーゼン・不規則な境界・高齢発症 |
| スターガルト病 | dark choroid・ABCA4変異 |
| 錐体ジストロフィ | 網膜電図錐体反応の著減・羞明が主症状 |
現時点でCACDに対する確立した治療法は存在しない。治療は症状の管理と生活の質の維持を目標とする。
ロービジョンリハビリ
拡大鏡・遮光眼鏡:残存視機能を最大限に活用するための補助具を処方する。
職業・生活訓練:視覚障害に対応した日常生活技術の習得を支援する。
遺伝カウンセリング
家族への情報提供:常染色体優性遺伝(50%の遺伝リスク)について説明する。
遺伝子検査:変異の同定は確定診断・将来の治療選択に重要である。
遺伝子治療(研究段階)
将来の治療候補:PRPH2変異を標的とした遺伝子補充療法の研究が進んでいる。
現状:現時点では一般医療として提供できる段階ではない。
PRPH2を含む遺伝性網膜疾患に対する遺伝子治療の研究は世界的に加速している。英国では脈絡膜ジストロフィ(コロイデレミア)に対する遺伝子治療試験が2014年に行われた実績があり、PRPH2変異疾患への応用も研究対象となっている。ただし現時点では標準治療としては確立されておらず、研究段階の段階である。
萎縮域では脈絡毛細管板・RPE・視細胞(桿体・錐体)が消失する。外顆粒層(ONL)の細胞数が著しく減少し、外境界膜(OLM)がブルッフ膜に直接接する状態となる。中大型の脈絡血管は病期の進行した段階でも長期間保持される。
ペリフェリン-2は視細胞外節ディスクの縁(rim領域)に局在し、ディスク形成・安定化・縁の湾曲維持に必須の役割を担う。
PRPH2ハプロ不全(1コピーの機能喪失)が生じると、正常な外節形態の形成が障害される。外節の構造破綻は視細胞アポトーシスへと進行し、RPEおよび脈絡毛細管板の二次的萎縮を招く。
PRPH2は桿体と錐体で異なる機能を担うことが示唆されており、PRPH2ノックアウトマウスモデルでは青色錐体が他の錐体より緩徐に変性することが報告されている。この桿体・錐体間の差異が疾患の臨床表現型の多様性に関与していると考えられる。
多局所網膜電図(mfERG)では、眼底検査で確認できる萎縮域を超えた傍中心窩でも機能低下が検出される。これはCACDの病変が臨床的に見えている萎縮よりも広範囲に及んでいることを示している。
PRPH2変異を含む遺伝性網膜疾患に対する遺伝子補充療法・遺伝子編集療法の研究が世界的に進められている。関連疾患(コロイデレミア)では2014年に英国で初の遺伝子治療臨床試験が行われた。
それぞれの変異の病原性解析がゲノム医療の進展とともに精緻化されている。全エクソーム解析(WES)の普及が診断精度を大幅に向上させており1)、より多くの患者の変異同定を可能にしている。
PRPH2が桿体と錐体で異なる機能的役割を持つことが分子レベルで解明されつつある。この知見は、表現型の多様性(桿体優位変性か錐体優位変性か)を説明する上で重要であり、将来の治療標的の選択に影響する可能性がある。