脈絡網膜ラクナ
分布:両側性。乳頭周囲・後極に密集するが、周辺部にも及ぶ。
外観:円形〜楕円形の黄白色〜ピンク色病変。有病率は70〜90%1)。
組織学:網膜色素上皮(網膜色素上皮)の欠損が脈絡膜層から裸の強膜層に至る2)。
経過:術後に経時的にサイズや数が増大する場合がある2)。

エカルディ症候群(Aicardi syndrome)は、1967年にフランスの神経科医Jean Aicardiが初めて記載した先天性の希少疾患である。X連鎖優性遺伝と推定され、ほぼすべての患者が女児である。男児ではヘミ接合型致死となるため、XXY核型(クラインフェルター症候群)の男児での報告は数例にとどまる。
発生率は約1:110,000出生と推定され、世界中での罹患者数はおよそ4,000人とされる1)。すべての症例がde novo変異であり、親から子への伝達例はなく、兄弟への再発リスクは1%未満である1)。
古典的三徴として以下の3つが知られる1)。
予後は不良である。平均生存年齢は18歳であり、27歳まで生存する確率は0.62%と報告されている1)。
本疾患はほぼ女児にのみ発症する。X連鎖優性遺伝と推定され、男児ではヘミ接合型で致死となるためである。ただしXXY核型(クラインフェルター症候群)を持つ男児での報告が世界で数例存在する。
本疾患の初発症状は通常、生後3〜4ヶ月頃に出現する点頭てんかんである。てんかんは薬剤抵抗性に移行することが多く、多様な発作型を伴う。
本疾患の眼科的所見のうち、脈絡網膜ラクナは本疾患に病的特異的(pathognomonic)な所見とされる。
脈絡網膜ラクナ
分布:両側性。乳頭周囲・後極に密集するが、周辺部にも及ぶ。
外観:円形〜楕円形の黄白色〜ピンク色病変。有病率は70〜90%1)。
組織学:網膜色素上皮(網膜色素上皮)の欠損が脈絡膜層から裸の強膜層に至る2)。
経過:術後に経時的にサイズや数が増大する場合がある2)。
その他の眼所見
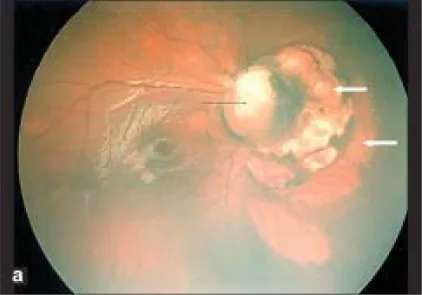
症例報告では、右眼に網膜前出血と360度周辺部無血管帯、左眼にstalk tissue(線維血管茎)と牽引性網膜剥離が認められた例が記録されている2)。また、皮質視覚障害(cortical visual impairment; CVI)の原因となり得る3)。
進行する場合がある。眼科的手術介入後にラクナが新たに可視化されたり、経時的にサイズや数が増大したりすることが報告されている2)。定期的な眼底検査による経過観察が重要である。
エカルディ症候群はX連鎖優性遺伝と推定されているが、原因遺伝子は現時点で特定されていない1)。全症例がde novo変異であり、家族内発症は基本的に見られない。兄弟への再発リスクは1%未満であり、遺伝カウンセリングの上で次子の計画を検討することが推奨される1)。
近年の症例報告では、TREX1遺伝子変異(c.292_293insA、p.(Cys99Metfs))が1例で検出された2)。TREX1は3番染色体上に位置し、X連鎖との仮説と一部矛盾するため、原因遺伝子の所在については議論が続いている。
エカルディ症候群の診断は、原因遺伝子が未特定であるため臨床診断が主体となる。以下の古典的三徴の確認が診断の中心となる1)。
三徴のうち2つしか満たさない場合でも、1999年に策定された拡張診断基準で診断が可能である。
拡張診断基準の構成を以下に示す。
| 分類 | 主な項目 |
|---|---|
| 主要特徴 | 視神経コロボーマ、皮質奇形、異所性灰白質、頭蓋内嚢胞、脈絡叢乳頭腫 |
| 支持的特徴 | 椎骨肋骨異常、小眼球症、分離脳EEG、半球非対称、血管奇形 |
三徴のうち2つ+主要特徴または支持的特徴が2つ以上で診断を確定できる。
現時点では確定診断可能な原因遺伝子は特定されていないため、遺伝子検査のみで確定診断はできない1)。臨床症状・眼底所見・画像所見を総合した臨床診断が主体となる。網羅的ゲノム解析の進歩により、将来的な原因遺伝子の特定が期待されている。
根治療法は存在しない。治療の目標は、てんかんコントロール・眼科合併症への対処・リハビリテーションによる発達支援の3本柱からなる。
てんかん管理
第一選択薬:フェニトイン、レベチラセタム、クロバザムなどを使用する1)。
難治例:カンナビジオール(CBD)・ケトン食療法・脳梁離断術・迷走神経刺激療法が試みられる1)。
眼科的介入
レーザー光凝固:周辺部無血管網膜に対して施行し、増殖性網膜症の進行を予防する2)。
硝子体切除術:牽引性網膜剥離(TRD)に対して23G硝子体切除術を施行する2)。
リハビリ
早期開始:診断直後から理学療法・作業療法・言語療法・視覚療法を開始する1)。
ロービジョンケア:視覚障害に対する補助器具や環境調整を行う。
眼科的合併症に対する手術介入の報告例を示す。
| 眼 | 所見 | 治療 |
|---|---|---|
| 右眼 | 網膜前出血・無血管帯360度 | レーザー光凝固 |
| 左眼 | stalk tissue・TRD | 23G硝子体切除術 |
硝子体切除術後には眼軸長の成長回復が確認されており、左眼において生後1ヶ月時17.45mmであった眼軸長が26ヶ月時には24.41mmまで成長した2)。また、術後に脈絡網膜ラクナが新たに可視化され、診断確定に寄与した例も報告されている2)。
可能である。周辺部無血管網膜に対するレーザー光凝固や、牽引性網膜剥離に対する23G硝子体切除術が有効な場合がある2)。手術後に眼球成長が促進された報告もある。ただし症例数が少なく、専門施設での慎重な対応が求められる。
X染色体上のde novo変異が本疾患の原因と推定されている。X染色体不活化(ライオニゼーション)のパターンにより、同一変異でも表現型に多様性が生じると考えられる。男性ではヘミ接合型となるため胎生期に致死となり、XXY核型を持つ男性のみが生存できる。
近年、TREX1遺伝子変異(c.292_293insA、p.(Cys99Metfs))が本疾患と診断された1症例で検出された2)。TREX1は3番染色体上に位置するため、X連鎖仮説と矛盾する可能性があり、非X連鎖の原因遺伝子が存在する可能性も示唆されている。
脈絡網膜ラクナの組織学的特徴として、網膜色素上皮(網膜色素上皮)の欠損が脈絡毛細血管板から裸の強膜層に至ることが確認されている2)。欠損部位には網膜色素上皮および脈絡毛細血管板が存在せず、未分化な神経網膜の異形成が認められる。
持続胎児血管系(persistent fetal vasculature)の残存が、線維血管茎(stalk tissue)や周辺部無血管網膜の形成に関与すると考えられている2)。これらの異常血管が牽引性網膜剥離の原因となる。
皮質視覚障害(CVI)は、多小脳回・異所性灰白質・脳梁欠損などの脳構造異常に起因する後天的な視覚機能低下として生じる3)。
脳奇形(多小脳回・脳梁欠損)は胎生期の神経細胞移動障害を基盤とする。この移動障害の原因となる分子メカニズムは未解明のままであり、原因遺伝子の特定とともに今後の研究が待たれる。
原因遺伝子は未特定のままであるが、TREX1遺伝子変異の検出2)は非X連鎖の変異が関与する可能性を示す新たな手がかりである。網羅的ゲノム解析(WES・WGS)の普及により、原因遺伝子の特定が加速されることが期待される。X連鎖仮説と非X連鎖仮説の検証は今後の主要課題である。
脳梁欠損が完全でなく菲薄化(dysgenesis)のみを示す変異型の報告が蓄積しつつある2)。三徴をすべて満たさない症例でも詳細な眼底検査と拡張診断基準の適用により診断精度が向上する可能性がある。
Kangら(2022)は、エカルディ症候群の両眼性硝子体網膜症症例に対してレーザー光凝固と23G硝子体切除術を施行し、眼球成長の維持と視覚機能の保護に寄与したことを報告した2)。また術後に脈絡網膜ラクナが新たに可視化されたことも記録されており、眼科的手術が診断確定にも貢献し得ることを示した。
早期の眼底スクリーニングと迅速な介入が網膜機能保護に重要であるとの認識が高まっている2)。周辺部無血管領域へのレーザーが増殖性網膜症の進行を予防する可能性があり、今後の症例集積が望まれる。
カンナビジオール(CBD)やケトン食療法のてんかんコントロールへの応用研究が進んでいる1)。難治性てんかんに対する新規治療選択肢の確立が期待される。